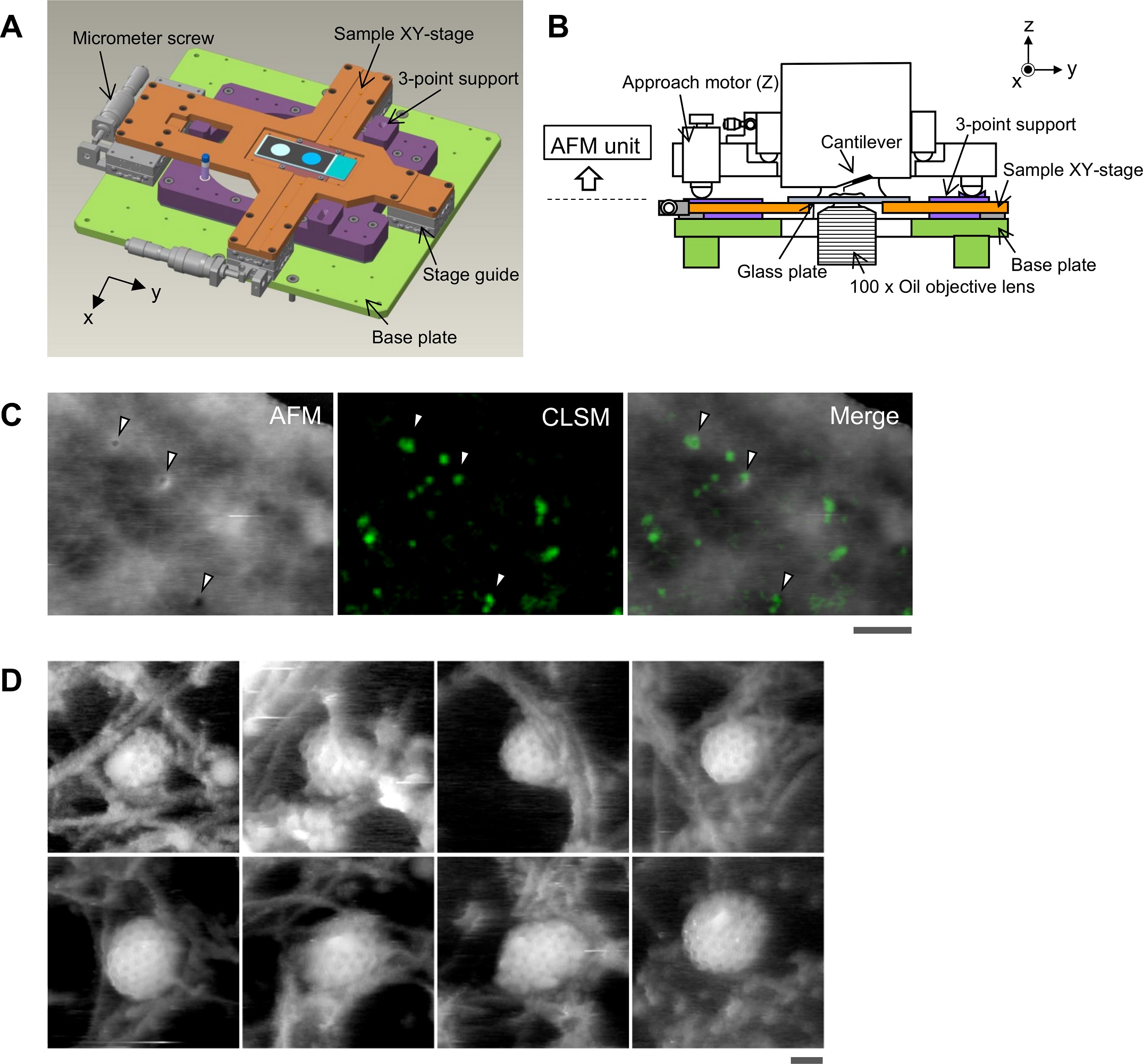
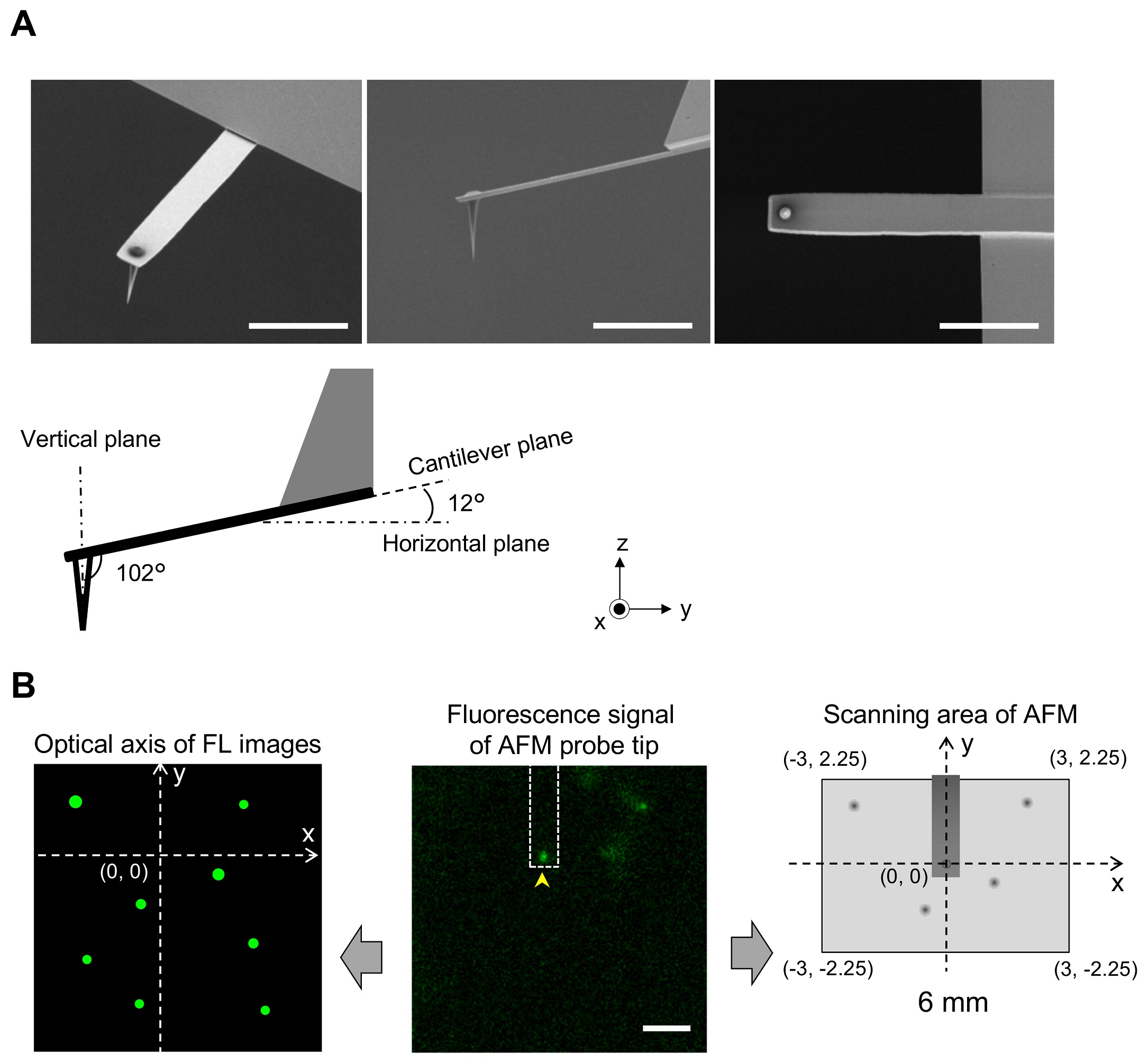
During replication, expression, and repair of the eukaryotic genome, cellular machinery must access the DNA wrapped around histone proteins forming nucleosomes. These octameric protein·DNA complexes are modular, dynamic, and flexible and unwrap or disassemble either spontaneously or by the action of molecular motors. Thus, the mechanism of formation and regulation of subnucleosomal intermediates has gained attention genome-wide because it controls DNA accessibility.*
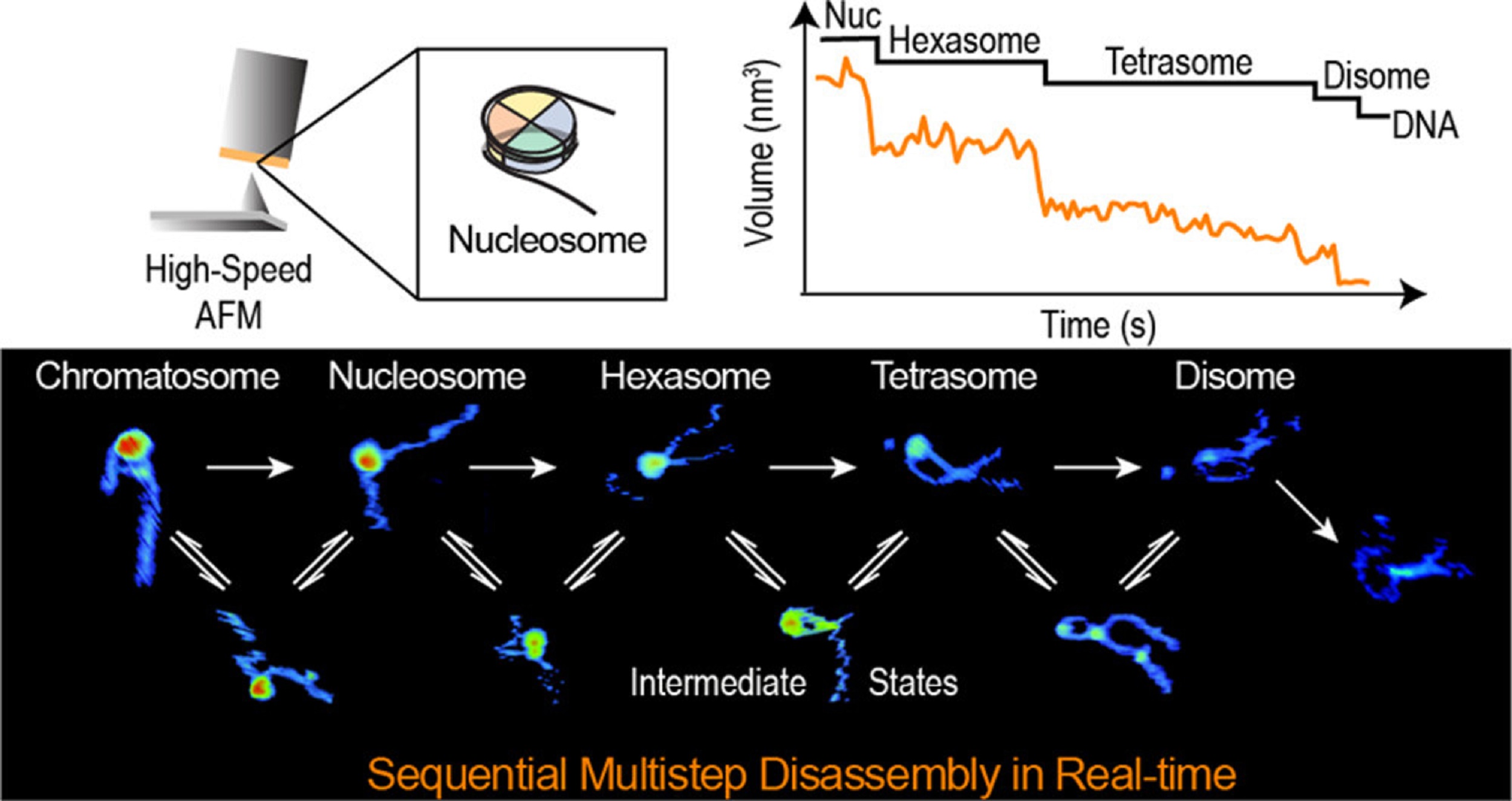
In the article “Real-Time Multistep Asymmetrical Disassembly of Nucleosomes and Chromatosomes Visualized by High-Speed Atomic Force Microscopy” Bibiana Onoa, César Díaz-Celis, Cristhian Cañari-Chumpitaz, Antony Lee and Carlos Bustamante describe how they imaged nucleosomes and their more compacted structure with the linker histone H1 (chromatosomes) using high-speed atomic force microscopy to visualize simultaneously the changes in the DNA and the histone core during their disassembly when deposited on mica.*
Furthermore, Bibiana Onoa et al. trained a neural network and developed an automatic algorithm to track molecular structural changes in real time. *
The authors’ results show that nucleosome disassembly is a sequential process involving asymmetrical stepwise dimer ejection events. The presence of H1 restricts DNA unwrapping, significantly increases the nucleosomal lifetime, and affects the pathway in which heterodimer asymmetrical dissociation occurs. *
Bibiana Onoa et al. observe that tetrasomes are resilient to disassembly and that the tetramer core (H3·H4)2 can diffuse along the nucleosome positioning sequence. Tetrasome mobility might be critical to the proper assembly of nucleosomes and can be relevant during nucleosomal transcription, as tetrasomes survive RNA polymerase passage. These findings are relevant to understanding nucleosome intrinsic dynamics and their modification by DNA-processing enzymes. *
To characterize the nucleosomes dynamics in 2D, individual molecules were observed in buffer using an Ando-type high speed atomic force microscope together with NanoWorld Ultra-Short Cantilevers for HS-AFM of the USC-F1.2-K0.15 AFM probe type ( typical spring constant 0.15 N/m, typical resonance frequency in air 1200 kHz, resonance frequency 500–600 kHz in liquid). *
The AFM data presented in the article allow the authors to directly visualize the dynamics of DNA and histones during nucleosome and chromatosome disassembly, providing a simultaneous observation of DNA unwrapping and histone dissociation. *
The experimental and analytical strategy presented shows that real-time HS-AFM is a robust and powerful tool for studying single nucleosomes and chromatin dynamics. *
*Bibiana Onoa, César Díaz-Celis, Cristhian Cañari-Chumpitaz, Antony Lee and Carlos Bustamante
Real-Time Multistep Asymmetrical Disassembly of Nucleosomes and Chromatosomes Visualized by High-Speed Atomic Force Microscopy
ACS Central Science 2024, 10, 1, 122–137
DOI: https://doi.org/10.1021/acscentsci.3c00735
Open Access The article “Real-Time Multistep Asymmetrical Disassembly of Nucleosomes and Chromatosomes Visualized by High-Speed Atomic Force Microscopy” by Bibiana Onoa, César Díaz-Celis, Cristhian Cañari-Chumpitaz, Antony Lee and Carlos Bustamante is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.